
































敗血症性ショックの病態は,時間経過とともにwarm shockからcold shockへ変化していく
敗血症性心筋症のメカニズムは明らかになりつつあるが,いまだ明確な結論は得られていない

敗血症性心筋症には2つのタイプが存在し,心筋収縮能が低下して心室拡張が認められるタイプは予後良好であるが,拡張障害を示すタイプは予後不良である
敗血症性ショックの初期には,血管内皮細胞から過剰に産生された一酸化窒素(NO)などにより血管抵抗が減弱した血液分布異常性ショックの様相を呈し,末梢は温暖であることからwarm shock,あるいは心拍出量が増加し高心拍出量性ショック(hyperdynamic state)とも呼ばれる病態を呈する。
しかし,敗血症性ショック患者の心機能は初期の段階から抑制されており1),末梢血管拡張がその病態をマスクしている。そのため,血管内皮細胞の損傷・脱落に伴い血管拡張物質の産生が低下すると,エンドセリン,トロンボキサンA2,アンジオテンシンⅡなどの血管収縮作用により後負荷が上昇し,低心拍出量性ショック(hypodynamic state),いわゆる末梢循環の損なわれたcold shockの病態を呈することになる。
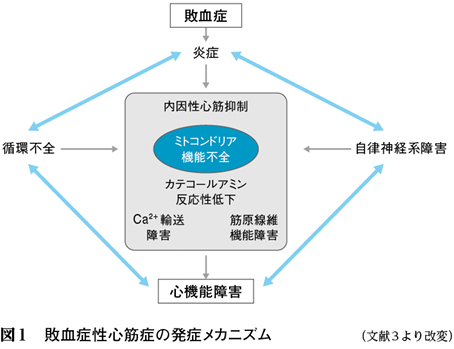
このような敗血症性ショックにおける心機能障害に対して,敗血症性心筋症(septic cardiomyopathy)2)という病態が提唱されている。
敗血症性心筋症の発症メカニズムのひとつに,β受容体のダウンレギュレーションがある。ラットを用いた実験では,エンドトキシンにより心筋のβ受容体の数が減少することが示され,また,β受容体情報伝達系における抑制性G蛋白の増加や間接的なprotein kinase A活性の抑制によるカテコールアミン反応性の低下も報告されている。敗血症の初期では,過剰に分泌されたNOがペルオキシナイトライトを形成し,L型Ca2+チャネル機能を抑制することや,細胞内ミトコンドリアの障害が引き起こされること,心筋細胞のアポトーシスが敗血症性心筋症に関与していることも指摘されている。さらに,筋小胞体に貯蔵してあるCa2+が枯渇し心筋収縮力の低下がみられ,逆に拡張期には筋小胞体からのCa2+の放出が促され,細胞内のCa2+濃度上昇に伴う拡張障害などを発症することが報告されている4)。これまでの多くの研究から敗血症性心筋症のメカニズムは明らかになりつつあるが,いまだ明確な結論は得られていない5)。

残り4,061文字あります
会員登録頂くことで利用範囲が広がります。 » 会員登録する








